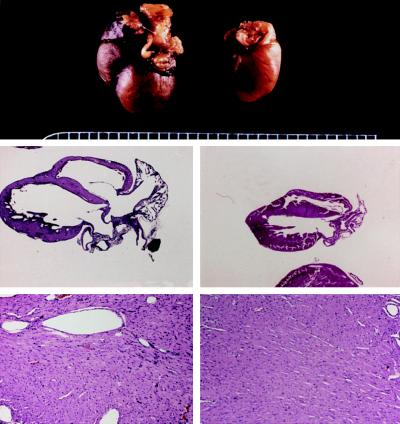
Neurohormonal stimulation by angiotensin II, endothelin, phenylephrine, and other hormones that activate Gq/protein kinase C signaling pathways is an essential mediator of cardiac hypertrophy. Accordingly, inhibition of angiotensin signaling with converting enzyme inhibitors, receptor blockers and aldosterone antagonists is standard therapy. Over a decade ago we were the first to establish the primacy of Gq/PKC signaling in cardiac hypertrophy by forcing activation of this pathway in the absence of neurohormonal activity through cardiac-specific overexpression of the alpha subunit of heterotrimeric Gq (Proc Natl Acad Sci USA 94:8121-8126, 1997) (Figure 1).
This model is now used by dozens of laboratories worldwide as the standard genetic model of pathological cardiac hypertrophy. Our follow-up studies used cardiac expression of peptide PKC translocation modifiers to define different roles for PKC isoforms, α, δ, and ε, in aspects of hypertrophy and contraction. Ongoing studies are utilizing conditional gene ablation models we created to examine the consequences of combined cardiomyocyte-specific ablation of novel PKCs δ and ε on the hypertrophic and apoptotic responses to pressure overload and myocardial infarction.
Figure 1.
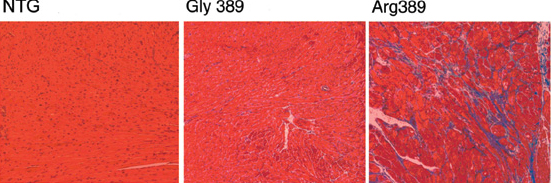
Activation of the sympathetic nervous system also contributes to cardiomyopathies by stimulating β-adrenergic receptors coupled to Gs/PKA signaling, as we described in mice overexpressing β-adrenergic receptors in the heart (Proc Natl Acad Sci USA 96:12798-12803, 1999; Circulation 101:1707-1714, 2000). Consequently, β-adrenergic receptor blockade is life-prolonging therapy, and we have found gene variants within adrenergic receptors (Nat Med 9:1300-5, 2003) (Figure 2) or the G-protein receptor kinases (GRKs) that desensitize them (Nat Med 14:510-7, 2008) that are associated with altered heart failure development, progression, or response to β-blocker therapy. Ongoing laboratory work aims to define the relative roles of cardiac-expressed GRKs 2, 5, and 6 individually and in combination using conditional gene ablation mouse models.
Figure 2.