Our interest in mitochondrial dynamics, mitochondrial pathways of programmed cell death, and mitochondrial quality control have led us from the heart to the brain, and specifically to create and investigate mouse models of Parkinson’s Disease.
Mitochondria are the essential ATP producers that drive homeostatic cell metabolism and fuel stress-responses. Because hearts have greater mitochondrial abundance and minute-by-minute requirement for ATP than other organs, they have been a useful model for interrogating in vivo mitochondrial function, defining novel ways in which mitochondria impact health or disease, and extrapolating these functions to other organs.
For example, by manipulating mitochondrial dynamics factors mitofusin and Opa, and mitochondrial quality control factor Parkin, in Drosophila heart tubes and mouse hearts, we uncovered novel mechanisms of cross-talk between the pathways that regulate mitochondrial fission and fusion, mitochondrial biogenesis, and mitochondrial elimination via mitophagy.
Thus, we determined that the mitochondrial fusion factor Mfn2 is phosphorylated by PINK1 to become the Parkin receptor that mediates mitophagy (Science. 340:471-5, 2013, pdf) and further established that mitochondrial fusion factors can help determine cell fate during heart differentiation (Science. 342:734-7, 2013, pdf).
We have advanced these studies into the brain using conditional deletion of Parkin and mitofusin genes in dopaminergic neurons of the substantia nigra and locus coeruleus, which are the main neurological sites of mitochondrial dysfunction and neuronal degeneration in Parkinson’s Disease.
Current efforts in the laboratory are focused on manipulating mitochondrial fusion to contain mitochondrial dysfunction in Parkin defective brains and hearts (Figure 1) (Circ Res. 114:257-65, 2014, pdf), which we hope will lead us to a new form of management for Parkinson’s Disease and other conditions caused by defective mitochondrial quality control.
Figure 1.
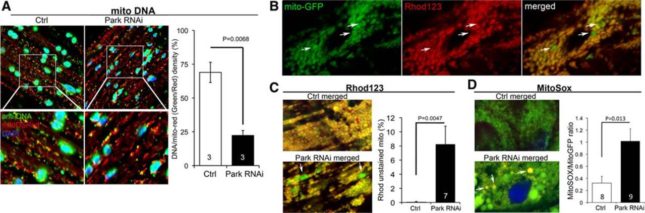
Cardiomyocyte mitochondrial dysfunction induced by Parkin suppression. (A) Merged confocal analysis of cardiomyocyte mitochondria nucleoids visualized using anti-DNA antibody (green) and cardiomyocyte-specific expression of Mito-DSRed (red). Nuclei visualized withDAPI (blue) were digitally masked and mitochondrial DNA content measured usingImageJ. Quantitative results are to the right. (B and C), Depolarization of structurally abnormal mitochondria assessed byrhodamine (Rhod) 123 staining. B, Separate tincΔ4-Gal4–drivenmito-GFP (green;left) andRhod123 (red;middle) and merged images (right) with representative depolarized (less red staining) mitochondria indicated with arrows. (C) Representative merged control and Parkin RNAimito-GFP/Rhod123 double-stained cardiomyocytes (left) and group quantitative data (right). Two of several depolarized mitochondria are indicated by arrows. (D) Representative merged control and Parkin RNAimito-GFP/MitoSoxdouble-stained cardiomyocytes (left) and group quantitative data (right). Three of several reactive oxygen species–producing mitochondria are indicated by arrows.